Calculating the pairwise RMSD of a trajectory¶
Last executed: Feb 06, 2020 with MDAnalysis 0.20.2-dev0
Last updated: January 2020
Minimum version of MDAnalysis: 0.17.0
Packages required:
Optional packages for data visualisation:
matplotlib
Note
MDAnalysis implements RMSD calculation using the fast QCP algorithm ([The05]). Please cite ([The05]) when using the MDAnalysis.analysis.align module in published work.
[1]:
import MDAnalysis as mda
from MDAnalysis.tests.datafiles import PSF, DCD
from MDAnalysis.analysis import diffusionmap, align
import matplotlib.pyplot as plt
%matplotlib inline
Loading files¶
The test files we will be working with here feature adenylate kinase (AdK), a phosophotransferase enzyme. ([BDPW09]) The trajectory DCD samples a transition from a closed to an open conformation.
[2]:
u = mda.Universe(PSF, DCD) # closed AdK (PDB ID: 1AKE)
Pairwise RMSD¶
Pairwise RMSDs are an effective way to quickly view similarities and differences in conformations (as measured by RMSD) across a trajectory.
First, you need to align the trajectory.
[3]:
aligner = align.AlignTraj(u, u, select='name CA',
in_memory=True).run()
We can then calculate a pairwise RMSD matrix with the diffusionmap.DistanceMatrix class, by using the default the rms.rmsd metric.
[4]:
matrix = diffusionmap.DistanceMatrix(u, select='name CA').run()
Step 98/98 [100.0%]
The results array is in matrix.dist_matrix as a square array with the shape (#n_frames, #n_frame).
[5]:
matrix.dist_matrix.shape
[5]:
(98, 98)
We can use the common plotting package matplotlib to create a heatmap from this array.
[6]:
plt.imshow(matrix.dist_matrix, cmap='viridis')
plt.xlabel('Frame')
plt.ylabel('Frame')
plt.colorbar(label='RMSD (Angstrom)')
[6]:
<matplotlib.colorbar.Colorbar at 0x11bccbeb8>
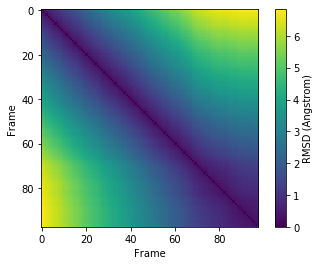
References¶
[1] Oliver Beckstein, Elizabeth J. Denning, Juan R. Perilla, and Thomas B. Woolf. Zipping and Unzipping of Adenylate Kinase: Atomistic Insights into the Ensemble of Open↔Closed Transitions. Journal of Molecular Biology, 394(1):160–176, November 2009. 00107. URL: https://linkinghub.elsevier.com/retrieve/pii/S0022283609011164, doi:10.1016/j.jmb.2009.09.009.
[2] Richard J. Gowers, Max Linke, Jonathan Barnoud, Tyler J. E. Reddy, Manuel N. Melo, Sean L. Seyler, Jan Domański, David L. Dotson, Sébastien Buchoux, Ian M. Kenney, and Oliver Beckstein. MDAnalysis: A Python Package for the Rapid Analysis of Molecular Dynamics Simulations. Proceedings of the 15th Python in Science Conference, pages 98–105, 2016. 00152. URL: https://conference.scipy.org/proceedings/scipy2016/oliver_beckstein.html, doi:10.25080/Majora-629e541a-00e.
[3] Naveen Michaud-Agrawal, Elizabeth J. Denning, Thomas B. Woolf, and Oliver Beckstein. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. Journal of Computational Chemistry, 32(10):2319–2327, July 2011. 00778. URL: http://doi.wiley.com/10.1002/jcc.21787, doi:10.1002/jcc.21787.
[4] Douglas L. Theobald. Rapid calculation of RMSDs using a quaternion-based characteristic polynomial. Acta Crystallographica Section A Foundations of Crystallography, 61(4):478–480, July 2005. 00127. URL: http://scripts.iucr.org/cgi-bin/paper?S0108767305015266, doi:10.1107/S0108767305015266.